Proteins are first cleaved into peptides by means of proteolytic enzymes such as trypsin. These peptides are separated by liquid chromatography and analyzed with mass spectrometry (LC-MS/MS). The spectra obtained by mass spectrometry are correlated to amino acid sequences, which on their turn are compared with databases from different kind of species (bacteria, human, mice, …).
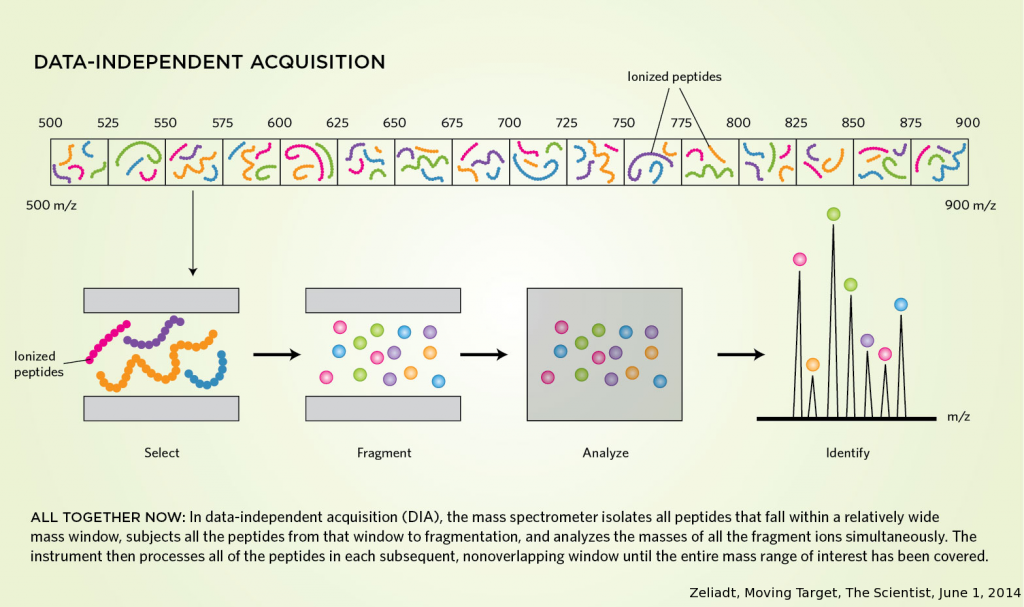
At ProGenTomics, protein identification can be performed on a TripleTOF5600 (Sciex) or on a SynaptG2Si (Waters) by both data-dependent acquisition (DDA) and data-independent acquisition (DIA). DDA is currently the most widespread strategy for protein identification where the most abundant precursors are selected for fragmentation. In DIA, on the contrary, all eluting peptides are fragmented which makes DIA an unbiased, reproducible quantitation method.
By means of an in-house developed method, the identity of a biological matrix and the species of forensic samples can be determined as well. This approach has the advantage that no extra trace material is needed since the analysis is performed on the first “washing” step of the DNA-extraction, a solution which is normally discarded, and that one single test is sufficient to determine the identity and the species of the biological matrix, while the conventional methods require cascade testing (Van Steendam et al., 2012, Int J Legal Med.).
Advantages of DDA
- Manual inspection of the spectra is possible
- Gold standard for protein identification
- Straightforward data analysis
Advantages of DIA
- All peptides get fragmented
- High reproducibility which is necessary for quantification
Workflow
You provide us with a protein sample, free from salts, preferably with a known amount. The proteins will first be digested into peptides by means of an enzyme. For quantification purposes, internal standards can be added to the sample. Peptides are first separated by means of hydrophobicity on a C18 column and subsequently analysed by a mass spectrometer (either TripleTOF 5600, Sciex or Synapt G2Si, Waters). The spectra will be searched against a database of choice and protein identification will be obtained after data-analysis with Mascot Daemon, Progenesis or DIANN.